
Peter Mayr Straße 1
6020 Innsbruck
Fax: +43 (0)512 9003 73502
Email: Johannes.Zschocke@i-med.ac.at
Website: https://www.i-med.ac.at/humgen/
Research year
Research Branch (ÖSTAT Classification)
301301, 301303, 301304, 304002
Keywords
Biochemical Genetics, Dental genetics, Ehlers-Danlos syndromes, Human Genetics, inherited metabolic disorders, Inherited tumour disposition syndromes, Lipidomics, Neurogenetics, Reproductive medicine, Syndromology, and Tumour genetics
Research Focus
Genetic causes of rare diseases and syndromes, incl. inherited metabolic diseases, Ehlers-Danlos syndromes, dental genetics, neurogenetics, reproductive genetics, developmental disorders, and others.
Genetic causes of tumours and tumour dispositions, incl. inherited cancer disposition syndromes, hamartomatous tumours, haematological malignancies.
Membrane lipid metabolism of organelles and cells.
Transcription and transcript processing, splicing.
New genetic laboratory methods, genotyping arrays, methylome analysis, mutation databases, quality control.
Genetic counselling.
Humangenom Austria project
General Facts
The primary aim of the Institute of Human Genetics is to clarify the genetic determinants of health and disease in humans and to translate this knowledge into individual medical care. This is achieved by combining comprehensive clinical services and all relevant laboratory diagnostic techniques with basic research. The main focus is on rare diseases that are inherited as monogenic traits with expansion to polygenic disease risk factors. The institute includes the ”Centre for Medical Genetics Innsbruck”, which provides medical genetic services and genetic counselling for Western Austria with extensive outpatient clinics and inpatient consultation in Innsbruck and several regional hospitals. The diagnostic and research laboratories cover all relevant methods for molecular genetic (DNA, RNA, epigenetics) and cytogenetic (genomic) analyses, biochemical analyses with a focus on lipidomic and a wide range of functional and cell biology techniques. The institute is dedicated to interdisciplinary collaboration and shares research activities with a large number of cooperation partners.
Research
On the 200th anniversary of Gregor Mendel’s birthday in 2022, we published an essay on the concepts of dominant and recessive for understanding inheritance, and the differences in the definition of these terms by Mendel and in genetic medicine. A subsequent major review article examined the different quantitative and qualitative effects of genetic variants and their relevance to understanding and treating inherited monogenic diseases.
Humangenom Austria: In 2023 we decided to organise the Austrian participation in the Genome of Europe project, a large pan-European project aimed at creating a European reference genome as part of the 1+ Million Genome Initiative. The project, which officially began at the Gregor-Mendel day (March 8th) 2025 with the kick-off meeting in Innsbruck, is carried out in collaboration with the Medical Universities Graz and Vienna and the Centre for Molecular Medicine Vienna under the lead (covering >50 % of the costs) of the Medical University Innsbruck. It will involve the initiation of a representative cohort study in the Austrian population in collaboration with the Institute of Clinical Epidemiology (Prof. Peter Willeit). One major aim is the establishment of a genome computing centre (lead scientist: Dr. Vincent Beliveau) with the appropriate genome analysis pipeline in Innsbruck (lead clinical laboratory geneticist: Martina Witsch-Baumgartner), the implementation of structured databases for genetic variants and diagnoses (cooperation with Prof. Peter Krawitz, Bonn) as well as a rare disease registry (cooperation with Dr. Georg Göbel, MUI). the comprehensive evaluation of the relevant ethical, legal and societal aspects for Austria is a major challenge (lead scientist: Dr. Michaela Th. Mayrhofer). The establishment of the genome network is expected to support the provision of medical genetic services in the public domain throughout Austria and provide extensive data for population-based genomic research.
European Reference Networks for Rare Diseases: In 2019, the institute was appointed Associated Centre of four European Reference Networks (ERNs): GENTURIS for genetic tumour risk syndromes (Lead: Katharina Wimmer), ReCONNET for connective tissue and musculoskeletal diseases (Lead: Johannes Zschocke), EURO-NMD for neuromuscular diseases (Lead: Sabine Rudnik), and ITHACA for congenital malformations and rare intellectual disability (Lead: Christine Fauth). Since 2024, the MUI represented by the Institute of Human Genetics is Austrian Expertise Centre (Type B Centre) for hereditary tumour disposition syndromes. In addition, the institute participates in several other Associated Centres established at the MUI and co-directs the Centre for Rare Diseases Innsbruck. Many rare diseases have a primary genetic origin, and medical genetics plays a major role in diagnosis, management and research of these conditions. This requires extensive multi-disciplinary networking.
Inherited Metabolic Disorders and Ehlers-Danlos Syndromes
Johannes Zschocke, Markus Keller, Sabrina Sailer, Gunda Schwaninger, Martina Witsch-Baumgartner, Albert Amberger
We focus on the diagnosis and the characterisation of clinical, biochemical and molecular features of inherited metabolic disorders, including various aspects of metabolism such as lipids, amino acids and organic acids. We developed the International Classification of Inherited Metabolic Disorders (ICIMD), a comprehensive collection of over 1,400 monogenic diseases. This classification is endorsed by international societies for inherited metabolic diseases and serves as the basis for the widely used handbook, the 5th Edition of the Vademecum Metabolicum and the recently released Online Inherited Metabolic Disorders WebApp (OIMD.org).
Familial Hypercholesterolaemia: Over several years, we have been investigating monogenic and polygenic causes of Familial Hypercholesterolaemia (FH) in Austria in cooperation with the University Hospital for Internal Medicine I at MUI. The identification of genetic scores has been established using the DNA array technology (Illumina iScan) in the institute in close collaboration with the Institute of Genetic Epidemiology at MUI (Prof. Florian Kronenberg and colleagues). Integrating genetic scores for LDL-C and Lp(a) increases the proportion of hypercholesterolemia patients with a clearly defined disease aetiology to 68.8% compared to 46.6% in standard genetic testing.
Ehlers-Danlos Syndromes: We have a long history in the identification and functional characterisation of monogenic causes of Ehlers-Danlos syndromes (EDS), a group of inherited connective tissue disorders characterised by joint hypermobility, skin hyper-elasticity and the fragility of various tissues. In 2024, we published a selection of comprehensive articles on the clinical, genetic and non-genetic diagnosis of monogenic EDS types. The most extensive research focus is on elucidating the pathogenetic mechanism that causes periodontal EDS (pEDS). Extensive functional studies of pEDS-associated C1r variants showed that the central elements in the pathogenesis of pEDS is intracellular activation of C1r and C1s and extracellular presence of activated C1s independently of microbial triggers. Recently, we identified activated C1s (aC1s) as an enzyme that degrades collagen I in patient cells and in-vitro assays (Figure 1).
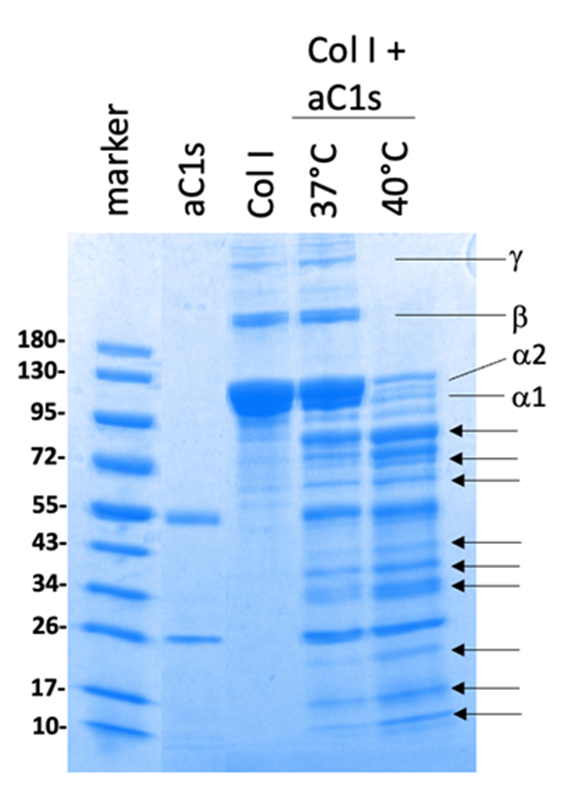
Collagen I (Col I) incubated without aC1s (lane 3) at 40°C overnight shows a weak smear below the 95 kDa band. At 37°C Col I is partial degraded, whereas at an incubation temperature of 40°C, high molecular weight Col I is almost completely degraded by aC1s (lanes 4 and 5). Degradation of Col I by aC1s resulted in various distinct Col I fragments as indicated by arrows.(Johannes Zschocke, Albert Amberger)
Interestingly, collagen I was completely degraded when assays were performed at 40°C, indicating that moderately elevated temperature has an impact on the integrity of collagen I. Degraded collagen I is expected to interfere with the compaction process that is important for mature tissue function, resulting in loosened tissue, a hallmark of the EDS phenotype. Our results indicate that pathogenesis in pEDS is mediated by inadequate activation of C1s and subsequent C1s-mediated degradation of matrix proteins, confirming pEDS as a connective tissue disorder.
Biochemical Genetics
Markus Keller
In our Biochemical Genetics Laboratory, we integrate genetic and multi-omics data using bioinformatics to understand genetically determined diseases. Our focus is on lipid metabolic processes and their impact on various diseases. We specialise in lipidomics, which is the study of how metabolic diseases affect membrane lipids and enzymatic processes. Our goal is to develop new therapies and analytical techniques for inherited metabolic disorders.
Regulation of Lipid Metabolism in Mitochondrial Membranes: Mitochondrial membrane lipid metabolism is vital for the optimal mitochondrial function. Dysregulation can cause inherited metabolic disorders, such as Barth syndrome (BTHS), characterised by severe cardiomyopathy. Changes in cardiolipin composition, a specific phospholipid, are observed in BTHS and various other conditions. Our recent studies have also uncovered a mechanistic link between cardiolipin metabolism and ferroptosis, further highlighting its importance in regulated cell death pathways.
Developing novel LC-MS/MS based lipidomics approaches: Accurate characterisation of complex lipid compositions is crucial for studying lipid metabolism, which is a dynamic, interconnected network sensitive to disease and nutrient status. Disturbances can alter both lipid class levels and fatty acyl compositions. However, identifying their origin is difficult due to shared metabolites, enzyme promiscuity and lipid diversity. LC-MS analysis at the molecular species level is further challenged by isobaric overlap, especially in complex lipids such as cardiolipin. We develop specialised LC-MS/MS techniques with optimised parameters and fragmentation strategies to identify and quantify challenging lipids such as cardiolipins, plasmalogens and ether lipids. These advancements enhance our understanding of their functional roles in cellular processes. We provide Fatty Acylizer (fatty-acylizer.lipid.at), a web tool that infers fatty acyl compositions from species-level data, using a combinatorial model and dynamic programming. It provides fast, reliable reconstruction, quality visualisations and easy integration into existing pipelines due to mzTab-M support. We hereby aim to support researchers in the study of remodelling lipid in health and disease.
Enzymology and evolutionary principles of HSD10: HSD10 is an essential mitochondrial protein with different functions and a significant evolutionary history. Mutations in HSD17B10 cause HSD10 disease, associated with cardiomyopathy and neurodegeneration. We demonstrated that previous reports on HSD10’s cardiolipin-cleaving activity are most likely an i- vitro artifact. As a crucial part of the mitochondrial RNase P complex, HSD10s evolutionary trajectory is constrained, thereby potentially impacting substrate specificity and reaction rates. Understanding these evolutionary principles is important for comprehending HSD10 substrate promiscuity and function.
Hereditary Cancer Genetics
Katharina Wimmer, Simon Schnaiter, Esther Schamschula
The mission of the Hereditary Cancer Genetics group is the improvement of cancer predisposition syndrome (CPS) diagnostics to identify novel genetic causes of CPS and to uncover the molecular mechanisms of tumorigenesis in patients with CPS.
Improvement of the molecular diagnostics of the Neurofibromatoses/Schwannomatoses and related disorders: In this project, we currently focus on the evaluation of the novel technique of long-read sequencing using PacBio technology to improve the diagnostics of NF1 as well as other CPS.
Genetic and clinical characterisation of constitutional mismatch repair deficiency (CMMRD) and related disorders: CMMRD caused by bi-allelic mutations in one of the four MMR genes is probably the most severe CPS and associated with a high risk for a broad spectrum of malignancies from early childhood onward throughout the entire life. Our and other’s work has led to improved understanding of this condition and the development of novel assays to assist in its diagnosis. Furthermore, existing surveillance protocols need adjustment considering recent observational prospective studies assessing their effectiveness. An update and merging of the different guidelines on diagnosis and clinical management of CMMRD into one comprehensive guideline was needed, also to include our current knowledge on the response of CMMRD-associated tumours to immune checkpoint inhibitors and the effectiveness and toxicity of other treatments. Therefore, under our lead, seventy-two experts of the European Reference Network GENTURIS and/or the European care for CMMRD consortium developed new guidelines on CMMRD diagnosis, genetic counselling, surveillance, quality of life and clinical management (Colas et al. 2024). In collaboration with two genetic centres in Belgium who provided patients and the Newcastle University which, in collaboration with us, had developed a highly reliable assay testing for the hallmark feature of CMMRD, i.e. constitutional microsatellite instability (cMSI), we were able to show that homozygosity or compound heterozygosity of hypomorphic MMR gene variants, frequently classified as variants of unknown significance, may cause an attenuated form of CMMRD that is associated with a phenotype reminiscent of Lynch syndrome, caused by heterozygous germline MMR gene pathogenic variants (PV). These findings not only have implications for diagnostics and clinical management of the affected patients, but also on our view on the current classification system of variants.
Genomic instability in ovarian cancer and other tumours: Genomic instability caused by homologous recombination deficiency (HRD) is a predictive biomarker for the response of different cancer types to PARP-inhibitor and platinum-based therapies. HRD results from the loss of function of genes involved in homologous recombination repair (HRR), but probably also from yet unknown mechanisms. The assessment of HRD, independent of the underlying variable causing the mechanism, can be performed by the detection of characteristic genomic instability resulting from the impaired HRR. As a part of our tumour profiling platform, we implemented and validated a simple and cost-effective method for the assessment of genomic instability. Next, among other methods, we will use machine learning to delineate the impact of genomic instability and copy number alterations on tumorigenesis, tumour phenotype and therapy response, including and beyond HRD in ovarian cancer and other tumour entities.
Genetic and Epigenetic Alterations in Haematological Malignancies and Solid Tumours
Emina Jukic, Simon Schwendinger, Verena Vogi
Since 2019, we have been working on genetic and epigenetic alterations in haematological malignancies and specific solid tumours. In addition, a specific focus of our working group clonal haematopoiesis (CHIP) in the context of various diseases.
Genetic alterations in haematological diseases: Together with the Austrian Myeloma Registry, Department of Internal Medicine II, Feldkirch and the Innsbruck University Hospital for Internal Medicine V, we focused on genetic alterations in diverse haematological diseases (Figure 2).
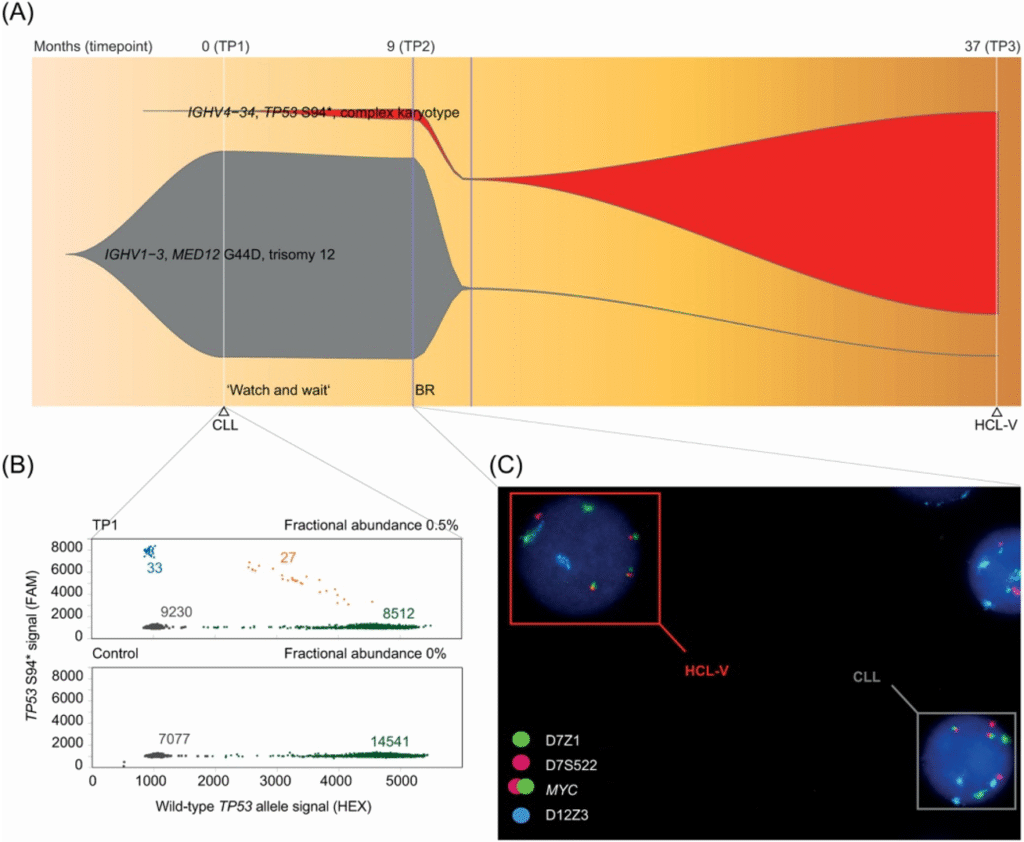
In multiple myeloma, we investigated chromosomal abnormalities and their impact on the course of the disease and therapy response. We identified amplification of chromosome 1q and tetraploidy as key genetic factors driving disease progression and therapeutic resistance. In our latest publication, we demonstrate the importance of genetic profiling in high-risk multiple myeloma for targeted therapies and improvement in outcomes. Additionally, we showed the genetic complexity of clonal dynamics in chronic lymphocytic leukaemia (CLL) and a hairy cell leukaemia-variant (HCL-v) leading to relapse and resistance. This emphasises the importance of understanding the genetic mechanisms underlying these diseases to enable accurate classification and personalised treatment approaches.
Genetic alterations in solid tumours: In addition to our work in haemato-oncology, in collaboration with the Visceral-Oncologic Centre at Ordensklinikum Linz, we are working on advancing liquid biopsy techniques for the genetic profiling of solid tumours. Gastrointestinal (GI) cancers, including gastroesophageal, colorectal and pancreatic cancers, serve as key models in our recent studies, where early ctDNA dynamics based on mutation and methylation specific analyses proved to be reliable predictors of treatment response. These findings highlight the relevance of genomic analyses in liquid biopsy for disease monitoring and treatment decisions.
Methylome analyses in oncologic diseases: In collaboration with the Institute of Medical Genetics and Pathology in Basel and the Institute of Human Genetics in Bonn, we investigate epigenetic alterations in different cancer type using DNA methylation array analysis to gain deeper biological insights into tumour heterogeneity. In our recent study on methylation patterns in cutaneous melanoma, conducted in cooperation with Innsbruck University Hospital for Dermatology, Venereology and Allergology, we showed distinct biological subgroups and revealed significant epigenetic differences compared to benign melanocytic nevi (Figure 3).
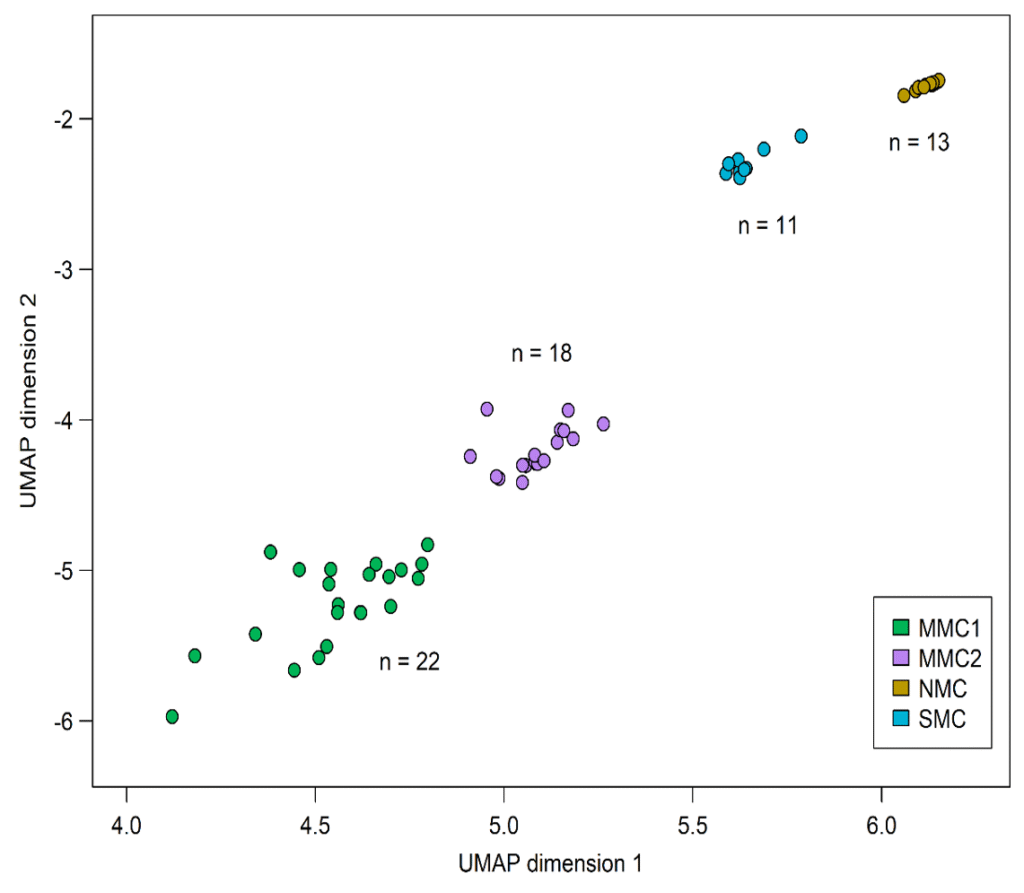
These findings highlight the potential of methylation-based classification not only for diagnostic refinement but also for disease stratification and deeper understanding of tumour biology. Based on this knowledge, our current research together with the Innsbruck University Hospital for Internal Medicine V focuses on methylation in myelodysplastic syndromes (MDS). The aim of this study is to combine epigenetic as well as genetic aberrations for a better classification and prognostication throughout the disease course. Furthermore, the result of this study should lead to an improvement of predicted prognosis in cases not fitting in the genetic scope.
Clonal Haematopoiesis of Indeterminate Potential in various diseases: With different research partners (Innsbruck University Hospitals for Internal Medicine II, III, IV and V and for Dermatology, Venereology and Allergology), we investigate CHIP (Clonal Haematopoiesis of Indeterminate Potential) including diabetic kidney disease, COVID-19, hematologic malignancies and atopic dermatitis, the impact of disease development and the progression as well as clinical course.
Neurogenetics
Sabine Rudnik, Roman Praschberger
The neurogenetic research in our institute is based on the long-term cooperation with partners of the EURO NMD network and the European Neuromuscular Centre. We have a particular interest in inherited peripheral neuropathies, neurogenic muscular atrophy, as well as undiagnosed hereditary myopathies and neurodevelopmental diseases. We helped to identify new genes or new genotype-phenotype correlations and have co-authored the updated guidelines on myotonic dystrophy and non-dystrophic myotonias in 2024. We made various research contributions regarding pregnancy and delivery in patients with neuromuscular disease. Since 2023, we are cooperating partners in a study on pregnancy and partnership in patients with spinal muscular atrophy, supported by the company Biogen.
In the newly formed Neurogenetics Laboratory, we study molecular and cellular mechanisms of neuronal vulnerability and resilience in rare neurogenetic disorders. For this purpose, we combine classical approaches such as genetic linkage analysis with short- and long-read next-generation sequencing in unresolved paediatric and adult patients with neurological phenotypes and perform causal disease modelling in experimental systems. To enable the rapid transition from variant discovery to in-vivo variant confirmation and mechanistic dissection, we make extensive use of the vast genetic toolkit in Drosophila.
Reproductive Genetics
Sabine Rudnik, Johannes Zschocke
For many years, we have actively participated in the development of guidelines in different areas of reproductive medicine. In 2021, the S2k guideline for couples with recurrent miscarriage was updated; the 3rd version is currently under revision (AWMF S2k-Leitlinie 015/050). The guideline on diagnosis and treatment prior to assisted reproduction (AWMF S2k-Leitlinie 015/85) is also currently under revision. The inconsistencies of international guidelines in terms of CFTR gene analysis in infertile men resulted in a multi-centre study of andrological findings in infertile men with two (biallelic) CFTR mutations. Moreover, we started an initiative to implement preconception carrier screening (PCS) in Austria. We hosted an international workshop on “Implementation of Preconception Carrier Screening in Austria” in November 2022 and published a report on the historical development and current status of PCS in German-speaking countries.
Genetic and Genomic Counselling
Gunda Schwaninger, Johannes Zschocke, Sabine Rudnik
The rapid development of genetic diagnostic methods leads to an increased need for competent professionals who can convey the background, goals, methods, results and consequences of genetic testing in the context of individual decision-making. MSc-trained Genetic Counsellors are widely integrated internationally, but we introduced the profession to the German-speaking countries only recently. Genetic Counsellors form a part of the inter-professional medical genetics team and complement patient care, particularly in psychosocial counselling aspects. In 2019, we established the first German-taught MSc programme in Genetic and Genomic Counselling at the MUI. The fourth 5-semester course begins in September 2025. In parallel, we are conducting research projects on the perception and optimal integration of this new profession within Austrian, German and Swiss genetic services. In June 2024, the German Society of Human Genetics established a commission for Genetic Counsellors with the goal to implement the profession in the human genetic services in the German-speaking countries. Further information is available at https://www.i-med.ac.at/gencouns/.
Pictures
Selected Publications
General topics:
- Zschocke J, Byers PH, Wilkie AOM. Mendelian inheritance revisited: dominance and recessiveness in medical genetics. Nat Rev Genet. 2023 Feb 20. doi: 10.1038/s41576-023-00574-0. Epub ahead of print. PMID: 36806206.
- Zschocke J, Byers PH, Wilkie AOM. Gregor Mendel and the concepts of dominance and recessiveness. Nat Rev Genet. 2022 Jul;23(7):387-388. doi: 10.1038/s41576-022-00495-4. PMID: 35508637.
Inherited Metabolic Disorders and Ehlers-Danlos syndromes:
- Schwaninger G, Forer L, Ebenbichler C, Dieplinger H, Kronenberg F, Zschocke J, Witsch-Baumgartner M: Filling the gap: Genetic risk assessment in hypercholesterolemia using LDL-C and LPA genetic scores. Clin Genet. 2023 104(3):334-343. PMID: 37417318 doi: [Text]
- Degradation of collagen I by activated C1s in periodontal Ehlers-Danlos Syndrome. Amberger A, Pertoll J, Traunfellner P, Kapferer-Seebacher I, Stoiber H, Klimaschewski L, Thielens N, Gaboriaud C, Zschocke J. Front Immunol. 2023 Mar 7;14:1157421. doi: 10.3389/fimmu.2023.1157421. PMC10028100.
- van Dijk FS, Angwin C, Ghali N, Zschocke J, Wagner B. Non-genetic diagnostic investigations in monogenic Ehlers-Danlos syndromes. Med Genet. 2024 Dec 3;36(4):247-254. doi: 10.1515/medgen-2024-2062. PMID: 39629472; PMCID: PMC11610441.
- Zschocke J, Demirdas S, van Dijk FS. Genetic diagnosis of the Ehlers-Danlos syndromes. Med Genet. 2024 Dec 3;36(4):235-245. doi: 10.1515/medgen-2024-2061. PMID: 39629471; PMCID: PMC11610442.
- van Dijk FS, Angwin C, Demirdas S, Ghali N, Zschocke J. Clinical diagnosis of the monogenic Ehlers-Danlos syndromes. Med Genet. 2024 Dec 3;36(4):225-234. doi: 10.1515/medgen-2024-2060. PMID: 39629459; PMCID: PMC11610443.
Biochemical Genetics:
- Keller, M. A. (2021). Interpreting phospholipid and cardiolipin profiles in rare mitochondrial diseases. Current Opinion in Systems Biology, 28(Icimd), 100383. https://doi.org/10.1016/j.coisb.2021.100383
- Koch, J., Watschinger, K., Werner, E. R., & Keller, M. A. (2022). Tricky Isomers—The Evolution of Analytical Strategies to Characterize Plasmalogens and Plasmanyl Ether Lipids. Frontiers in Cell and Developmental Biology, 10(April), 1–13. https://doi.org/10.3389/fcell.2022.864716
- Oemer, G., Edenhofer, M.-L., Wohlfarter, Y., Lackner, K., Leman, G., Koch, J., Cardoso, L. H. D., Lindner, H. H., Gnaiger, E., Dubrac, S., Zschocke, J., & Keller, M. A. (2021). Fatty acyl availability modulates cardiolipin composition and alters mitochondrial function in HeLa cells. Journal of Lipid Research, 62, 100111. https://doi.org/10.1016/j.jlr.2021.100111
- Oemer, G., Koch, J., Wohlfarter, Y., Lackner, K., Gebert, R. E. M., Geley, S., Zschocke, J., & Keller, M. A. (2021). The lipid environment modulates cardiolipin and phospholipid constitution in wild type and tafazzin-deficient cells. Journal of Inherited Metabolic Disease, n/a(n/a). https://doi.org/10.1002/jimd.12433
- Wohlfarter, Y., Eidelpes, R., Yu, R. D., Sailer, S., Koch, J., Karall, D., Scholl, S., Albert, B., Hauke, A., Zschocke, J., & Keller, M. A. (2022). Lost in promiscuity ? An evolutionary and biochemical evaluation of HSD10 function in cardiolipin metabolism. Cellular and Molecular Life Sciences, 1, 1–13. https://doi.org/10.1007/s00018-022-04579-6
Hereditary Cancer Genetics:
- BRCA loss of function including BRCA1 DNA-methylation, but not BRCA-unrelated homologous recombination deficiency, is associated with platinum hypersensitivity in high-grade ovarian cancer. Fiegl H*, Schnaiter S*, Reimer DU, Leitner K, Nardelli P, Tsibulak I, Wieser V, Wimmer K, Schamschula E, Marth C, Zeimet AG. Clin Epigenetics. 2024 Nov 27;16(1):171. doi: 10.1186/s13148-024-01781-0. PMID: 39605059 *equally contributing authors
- Stratification of Homologous Recombination Deficiency-Negative High-Grade Ovarian Cancer by the Type of Peritoneal Spread into Two Groups with Distinct Survival Outcomes. Schnaiter S, Schamschula E, Laschtowiczka J, Fiegl H, Zschocke J, Zeimet A, Wimmer K, Reimer D. Cancers (Basel). 2024 Jun 3;16(11):2129. doi: 10.3390/cancers16112129. PMID: 38893248
- ERN GENTURIS guidelines on constitutional mismatch repair deficiency diagnosis, genetic counselling, surveillance, quality of life, and clinical management. Colas C, Guerrini-Rousseau L, Suerink M, Gallon R, Kratz CP, Ayuso É; ERN GENTURIS CMMRD Guideline Group; Brugières L, Wimmer K. Eur J Hum Genet. 2024 Dec;32(12):1526-1541. doi: 10.1038/s41431-024-01708-6. Epub 2024 Oct 17. PMID: 39420201
- Constitutional mismatch repair deficiency mimicking Lynch syndrome is associated with hypomorphic mismatch repair gene variants. Gallon R, Brekelmans C, Martin M, Bours V, Schamschula E, Amberger A, Muleris M, Colas C, Dekervel J, De Hertogh G, Coupier J, Colleye O, Sepulchre E, Burn J, Brems H, Legius E, Wimmer K. NPJ Precis Oncol. 2024 May 24;8(1):119. doi: 10.1038/s41698-024-00603-z. PMID: 38789506
- Gonadal and gonadosomatic mosaicism in NF1: report of two families. Seidl-Philipp M, Veyt N, Schnaiter S, Krogsdam A, Schwendinger S, Maertens O, Fauth C, Schmuth M, Legius E, Wimmer K*, Brems H*. J Dtsch Dermatol Ges. 2024 Mar;22(3):426-428. doi: 10.1111/ddg.15302. Epub 2024 Jan 7. PMID: 38185792 *Equally contributing last authors. Ich
Genetic and Epigenetic Alterations in Haematological Malignancies and Solid Tumours:
- Locher M, Jukic E, Bohn JP, Untergasser G, Steurer M, Cramer CA, Schwendinger S, Vogi V, Verdorfer I, Witsch-Baumgartner M, Nachbaur D, Gunsilius E, Wolf D, Zschocke J, Steiner N. Clonal dynamics in a composite chronic lymphocytic leukemia and hairy cell leukemia-variant. Genes Chromosomes Cancer. 2021 Apr;60(4):287-292. doi: 10.1002/gcc.22925. Epub 2020 Dec 12. PMID: 33277788
- Petzer V, Schwendinger S, Haschka D, Vogi V, Tymoszuk P, Burkert F, Sahanic S, Sonnweber T, Bellmann-Weiler R, Loeffler-Ragg J, Tancevski I, Zschocke J, Weiss G, Wolf D, Jukic E. Clonal hematopoiesis in patients with Covid-19 is stable and not linked to an aggravated clinical course. Am J Hematol. 2021 Sep 1;96(9):E331-E333. doi: 10.1002/ajh.26251. Epub 2021 Jun 2. PMID: 34028864
- Denicolò S, Vogi V, Keller F, Thöni S, Eder S, Heerspink HJL, Rosivall L, Wiecek A, Mark PB, Perco P, Leierer J, Kronbichler A, Steger M, Schwendinger S, Zschocke J, Mayer G, Jukic E. Clonal Hematopoiesis of Indeterminate Potential and Diabetic Kidney Disease: A Nested Case-Control Study. Kidney Int Rep. 2022 Feb 3;7(4):876-888. doi: 10.1016/j.ekir.2022.01.1064. eCollection 2022 Apr. PMID: 35497780
- Vogi V, Haschka D, Forer L, Schwendinger S, Petzer V, Coassin S, Tancevski I, Sonnweber T, Löffler-Ragg J, Puchhammer-Stöckl E, Graninger M, Wolf D, Kronenberg F, Zschocke J, Jukic E, Weiss G. Severe COVID-19 disease is associated with genetic factors affecting plasma ACE2 receptor and CRP concentrations. Sci Rep. 2025 Feb 8;15(1):4708. doi: 10.1038/s41598-025-89306-4. PMID: 39922945
- Schwendinger S, Jaschke W, Walder T, Hench J, Vogi V, Frank S, Hoffmann P, Herms S, Zschocke J, Nguyen VA, Schmuth M, Jukic E. Diagnostics (Basel). 2025 Feb 21;15(5):531. doi: 10.3390/diagnostics15050531. PMID: 40075779. DNA Methylation Array Analysis Identifies Biological Subgroups of Cutaneous Melanoma and Reveals Extensive Differences with Benign Melanocytic Nevi.
Neurogenetics:
- Decet, M., Scott, P., Kuenen, S., Meftah, D., Swerts, J., Calatayud, C., Gallego, S.F., Kaempf, N., Nachman, E., Praschberger, R., Schoovaerts, N., Tang, C., Eidelberg, D., Al Adawi, S., Al Asmi, A., Nandhagopal, R., Verstreken, P. (2024). A candidate loss-of-function variant in SGIP1 causes synaptic dysfunction and recessive parkinsonism. Cell Reports Med. 5, 101749.
- Niebrügge, N., Trovato, O., Praschberger, R., and Lieb, A. (2025). Disease-Associated Dopamine Receptor D2 Variants Exhibit Functional Consequences Depending on Different Heterotrimeric G-Protein Subunit Combinations. Biomedicines 13.
- Kinnart, I., Manders, L., Heyninck, T., Imberechts, D., Praschberger, R., Schoovaerts, N., Verfaillie, C., Verstreken, P., and Vandenberghe, W. (2024). Elevated α-synuclein levels inhibit mitophagic flux. npj Park. Dis. 10, 1–15.
- Kaiyrzhanov R, … Rudnik-Schöneborn S, et al. (2024) Bi-allelic ACBD6 variants lead to a neurodevelopmental syndrome with progressive and complex movement disorders. Brain. 2024 147:1436-1456.
- Maroofian R, ….Rudnik-Schöneborn S,…. Zschocke J et al. (2023) Biallelic MED27 variants lead to variable ponto-cerebello-lental degeneration with movement disorders. Brain 146:5031-5043
Reproductive Medicine
- Rudnik-Schöneborn S, Zerres K (2022) Preconception carrier screeening as an alternative reproductive option prior to newborn screeening for severe recessive disorders. Med Genet 34:157-161
- Rudnik-Schöneborn S, Messner M, Vockel M, Wirleitner B, Pinggera GM, Witsch-Baumgartner M, Murtinger M, Kliesch S, Swoboda M, Sänger N, Zschocke J, Tüttelmann F (2021) Andrological findings in infertile men with two (biallelic) CFTR mutations: results of a multicentre study in Germany and Austria comprising 71 patients. Hum Reprod 36:551-559
Genetic and Genomic Counselling:
- Heidemann S, Zschocke J, & Schwaninger G. First experiences with the introduction of genetic counselors in human genetic services in the German-speaking countries. Journal of Genetic Counseling, 2024, 00, 1–11. https://doi.org/10.1002/jgc4.1979
- Schwaninger G, Heidemann S, Rudnik-Schöneborn S, Zschocke J. The current state of the Genetic Counselor Profession in the German-speaking Countries. Genetics in Medicine Open, 2024, 101849, https://doi.org/10.1016/j.gimo.2024.101849
- Schwaninger G, Taxer K, Marti S, Cionca S, Freilinger P, Gemperle C, Kalantidou A, Stöhr H, Ramcke S, Wölwer C, Rudnik-Schöneborn S, Zschocke J. Clinical Training and Integration of Genetic Counselors into Interprofessional Teams in the German-speaking Countries. Genetics in Medicine Open, 2024, 101855, https://doi.org/10.1016/j.gimo.2024.101855
Selection of Funding
- FWF P33333 to Markus Keller
- FWF P34574 to Markus Keller
- FWF I 6477-B to Katharina Wimmer
- FFG 878654 to Markus Keller
- DOC-Fellowship (ÖAW) to Jakob Koch
- DOC-Fellowship (ÖAW) to Yvonne Wohlfarter
- Digital Europe 101168231-GoE (EU) and FFG to Johannes Zschocke
Collaborations
- Daltro Cardoso Luiz Helena, Gnaiger Erich, Oroboros Instruments, Innsbruck, Austria
- Hauke S. Hillen, Ryan Yu, Max Planck Institute for Multidisciplinary Sciences and University of Göttingen, Germany
- Richard Gallon, Newcastle University, Newcastle, UK
- Nicole Thielens, Christine Gaboriaud, Univ. Grenoble Alpes, Institut de Biologie Structurale (IBS), Grenoble, France.
- Alban Lermine, BNDMR, MOABI-APHP and SeqOIA, Paris, France
- Peter Krawitz, Institute for Genomic Statistics and Bioinformatics, University of Bonn, Germany